

| Sommaire BO courant | Archives BO | Table des matières cumulée BO | Sommaire RMLR |
![]()
Instruction de procédure no 980034BPC du 30 octobre 1998 de suivi des protocoles de recherche biomédicale impliquant l'expérimentation avec l'homme
Bureau de pilotage et de coordination ; Sciences de la vie
Objet de l'instruction : la présente instruction examine les modalités d'application aux unités du CNRS, lors de la mise en oeuvre d'un protocole de recherche biomédicale impliquant l'expérimentation avec l'homme, de la loi du 20 décembre 1988 modifiée, dite loi Huriet-Sérusclat.
Personnes concernées : toute personne exerçant au sein des structures de recherches et de services du CNRS, quels que soient son statut et son organisme d'appartenance.
Période d'application : à compter du 1er décembre 1998
Adresse Web : http://www.sg.cnrs.fr/bpc
Dernière mise à jour : 30 octobre 1998
Coordonnées : Secrétariat général - Bureau de pilotage et de coordination
3, rue Michel-Ange - 75794 Paris Cedex 16.
Mél. : BPC.procedures@cnrs-dir.fr
Ce document a été établi en liaison avec
le département des sciences de la vie et la direction des contrats et des affaires
juridiques.
Mél. respectifs pour tout renseignement relatif aux règles de gestion, textes et
documents applicables : ethique.sdv@cnrs-dir.fr et DCAJ.procedures@cnrs-dir.fr.
La loi du 20 décembre 1988 modifiée, dite loi Huriet-Sérusclat, et son décret d'application du 20 septembre 1990, définissent le cadre de réalisation d'une recherche dans le domaine biomédical et ont surtout pour but de protéger les personnes qui se prêtent à ces recherches.
L'applicabilité des textes mais aussi leur complexité de mise en oeuvre nécessitent le recours quasi systématique à l'assistance de la cellule éthique du département des sciences de la vie. De plus, les instances réglementaires jugent indispensable un promoteur unique pour le CNRS. Ces deux raisons, l'origine et le nombre de dossiers soumis chaque année, ont conduit à revenir sur les options de déconcentration prises dans le passé.
Ainsi la présente instruction de procédures, qui abroge l'instruction no 910314SJUR du 19 novembre 1991 à laquelle elle se substitue, maintient au siège les décisions qui engagent la responsabilité de l'établissement. Le département des sciences de la vie est le représentant unique de l'organisme par délégation du directeur général. L'ensemble des modèles et formulaires (dont certains obligatoires) nécessaires aux investigateurs et aux délégations pour l'exécution de la procédure figurent en annexe.1
1. - PRÉSENTATION GÉNÉRALE, LES ACTEURS IMPLIQUÉS
La loi définit comme recherche biomédicale, « les essais ou expérimentations organisés et pratiqués sur l'être humain en vue du développement des connaissances biologiques ou médicales ».
Elle interdit ces recherches sur l'être humain :
« - si elles ne se fondent pas sur le dernier état des connaissances scientifiques et sur une expérimentation préclinique suffisante ;
- si le risque prévisible encouru par les personnes qui se prêtent à la recherche est hors de proportion avec le bénéfice escompté pour elles ou l'intérêt de cette recherche ;
- si elles ne visent pas à étendre la connaissance scientifique de l'être humain et les moyens susceptibles d'améliorer sa condition. »
Pour l'exposé des types de recherches non couverts par ce texte ou plus généralement pour tout problème d'interprétation du champ d'application, se référer au texte Recherche avec l'homme dans le dossier Éthique en sciences de la vie sur le Web du département SDV.
Les dispositions d'application diffèrent selon la catégorie de recherche biomédicale : avec ou sans bénéfice individuel direct pour la personne qui s'y prête, possibilité ou non d'effectuer ces recherches sur certaines catégories de personnes dites protégées (mineurs, majeurs sous tutelle, personnes privées de liberté...), possibilité ou non de les indemniser.
Dans tous les cas, la loi distingue principalement trois acteurs : le promoteur qui a l'initiative et la responsabilité de la recherche, l'investigateur qui dirige et surveille la recherche, et le sujet qui se prête à la recherche. Elle s'articule autour des principes suivants :
- Les lieux d'expérimentation doivent avoir été préalablement agréés pour ce type de recherche par le ministère chargé de la santé.
- L'investigateur doit être docteur en médecine et inscrit à l'ordre des médecins.
- Le protocole de recherche doit être soumis au ministère chargé de la santé après avis du CCPPRB ( cf . page 36).
- L'investigateur doit recueillir le consentement du sujet après l'avoir informé sur la nature, le déroulement et les risques de la recherche. Ce consentement peut dans certains cas être différé et recherché auprès de tiers autorisés ( cf. chapitre 4). De même, à titre exceptionnel, certaines informations telles que le diagnostic, ou le protocole d'expérience (dans le cas de recherches sur le comportement psychologique), peuvent ne pas être communiquées tout de suite au sujet dans son intérêt ou celui du protocole.
- Les contraintes subies par le sujet lors de recherches sans bénéfice individuel direct peuvent être indemnisées.
- Les éventuels dommages sur les personnes (décès, hospitalisation, séquelles) doivent être indemnisés. Le promoteur de la recherche a obligation de souscrire une assurance à cet effet.
Le ministre chargé de la santé et plus particulièrement la direction générale de la santé (DGS), ou l'agence du médicament, agréent les lieux d'expérimentation et les CCPPRB ( cf. infra ). Elles sont respectivement informées des protocoles de recherche entrepris qui les concernent et peuvent s'y opposer.
La direction régionale des affaires sanitaires et sociales (DRASS), et plus particulièrement le médecin inspecteur de la santé et le pharmacien inspecteur de la santé placés auprès d'elle, veillent au respect des dispositions de la loi. Ils reçoivent les demandes d'agrément des lieux d'expérimentation et rédigent un rapport après visite des locaux.
Le comité consultatif de protection des personnes dans la recherche biomédicale (CCPPRB) est saisi par l'investigateur et donne son avis sur les conditions de validité de la recherche au regard de la protection des personnes. Sa création et sa compétence géographique (généralement régionale) sont fixées par arrêté du ministre chargé de la recherche.
La Commission nationale de l'informatique et des libertés (CNIL) doit être saisie par l'intermédiaire de la direction des contrats et des affaires juridiques du CNRS (DCAJ), dès lors que le protocole de recherche nécessite que des données nominatives soient informatisées.
Le promoteur de la recherche est la personne physique ou morale (ici le CNRS, aux conditions exposées chapitre 4) qui prend l'initiative et la responsabilité de la recherche. Il doit avoir souscrit une assurance responsabilité civile.
Le département scientifique valide le projet de recherche sur le plan scientifique. Il sollicite l'accord du partenaire pour que le CNRS soit promoteur lorsque l'investigateur n'est pas affecté dans une unité propre du CNRS.
Le département SDV assure pour le CNRS les obligations de promoteur : paiement du droit fixe au CCPPRB, cotisation d'assurance responsabilité civile. Son directeur signe par délégation du directeur général, les demandes d'avis au CCPPRB et les déclarations envoyées au ministère.
La cellule éthique du département SDV conseille l'investigateur et suit au plan administratif et éthique les protocoles en cours. Elle soumet au Comité opérationnel pour l'éthique dans les sciences de la vie du CNRS (COPE) les demandes d'avis au CCPPRB.
La délégation du siège assure les relations avec la compagnie d'assurance et comptabilise à ce titre les déclarations de mise en oeuvre. Elle acquitte le droit fixe pour chaque dossier présenté au CCPPRB.
L' investigateur est la personne qui dirige, surveille, et éventuellement réalise la recherche. C'est obligatoirement un docteur en médecine inscrit à l'Ordre des médecins. Dans les sciences du comportement humain, une personne qualifiée peut conjointement avec l'investigateur, exercer la direction de la recherche. Lorsque l'investigateur n'est pas le directeur de l'unité, il adresse les documents au CNRS sous couvert de son directeur.
Le titulaire de l'autorisation des lieux est la personne physique (généralement l'investigateur), docteur en médecine, qui a rédigé la demande d'agrément.
Le responsable des lieux est la personne morale propriétaire ou attributaire des locaux où se déroulent les recherches. Pour les locaux CNRS, le délégué régional signe la demande d'autorisation des lieux adressée à la DRASS.
La délégation et plus particulièrement l'inspecteur régional d'hygiène et sécurité sont informés lorsqu'une recherche biomédicale est mise en oeuvre dans des locaux CNRS sous sa responsabilité. Elle rembourse le cas échéant au sujet les frais engagés et lui verse les indemnités pour contraintes subies.
Le sujet se prêtant à une recherche biomédicale est informé : de l'objectif de la recherche, de la méthodologie, de la durée, des bénéfices attendus, des contraintes, des risques prévisibles, de l'avis du CCPPRB, de son inscription sur le fichier national géré par le ministère. Il donne son consentement et peut le révoquer à tout moment.
2. - LE CIRCUIT DES DOCUMENTS, LES TÂCHES PAR ACTEUR
Ce chapitre traite du cheminement des documents ainsi que des tâches exécutées au sein de chaque entité.
Principaux qualificatifs utilisés :

2.1. Procédure : Agrément des lieux CNRS d'expérimentation
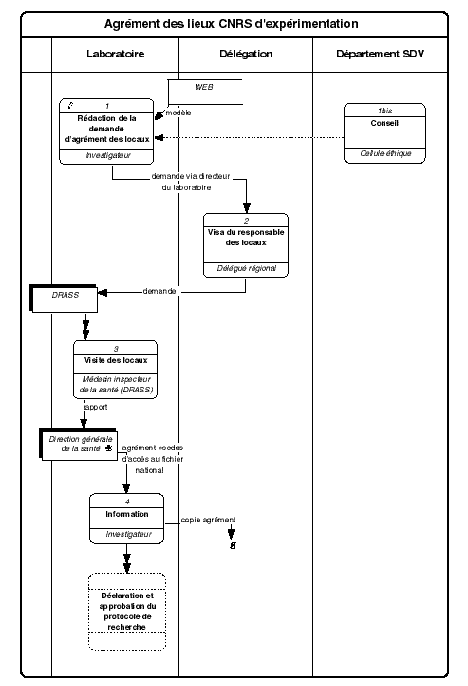
Selon la loi, les recherches impliquant l'expérimentation avec l'homme doivent être réalisées dans des locaux agréés. L'autorisation est attribuée à une personne physique et est valable pour un(des) type(s) de recherche bien défini(s). Toute modification de l'un de ces paramètres doit entraîner le renouvellement de la demande d'agrément. La demande doit être visée par la personne morale responsable des locaux.
L' investigateur qui souhaite effectuer des recherches biomédicales impliquant l'expérimentation sur l'homme dans des locaux dont le CNRS a la responsabilité, rédige une demande d'agrément de ces locaux. Un modèle est disponible en annexe.2 Il adresse la demande sous couvert de son directeur d'unité à la délégation. La cellule éthique du département SDV peut être consultée.
Le délégué régional vise la demande et l'adresse à la direction régionale des affaires sanitaires et sociales (DRASS).
Le médecin inspecteur régional de la santé (éventuellement le pharmacien inspecteur de la santé) auprès de la DRASS visite les locaux et vérifie leur conformité en fonction du type des expériences qui y seront menées. Un rapport est adressé à la direction générale de la santé (DGS).
Celle-ci retourne à l'investigateur l'agrément des lieux ainsi que les codes d'accès au fichier national des sujets d'expérimentation. L' investigateur transmet une copie de cet agrément à la délégation.
2.2. Procédure : Déclaration et approbation du protocole de recherche
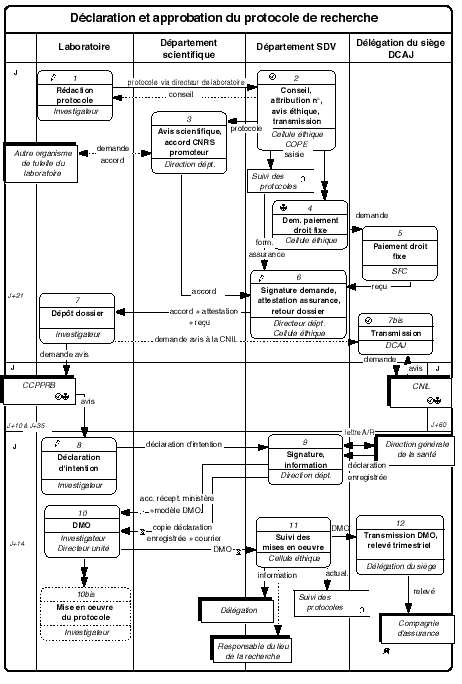
[voir représentation graphique page 41]
L' investigateur décrit le protocole de recherche en rédigeant la demande d'avis au comité consultatif de protection des personnes dans la recherche biomédicale (CCPPRB) selon le modèle fourni par la cellule éthique du département des sciences de la vie et disponible en annexe3 . Le dossier accompagné d'une copie de l'autorisation des lieux est envoyé au département SDV par l'intermédiaire de son directeur d'unité.
La cellule éthique du département des sciences de la vie attribue au protocole un numéro d'enregistrement qui permettra le suivi (année + no d'ordre) et saisit les éléments du dossier dans une base de suivi. Elle vérifie la conformité du dossier et sollicite l'avis du COPE (Comité opérationnel pour l'éthique dans les sciences de la vie du CNRS). Le protocole est adressé au département dont relève l'investigateur.
Le département scientifique évalue le protocole sur le plan de l'opportunité scientifique. Lorsque l'investigateur n'est pas affecté dans une unité propre du CNRS, le département sollicite l'accord du partenaire en vue d'accepter que le CNRS soit promoteur de la recherche (sauf dans les cas où cette clause figure dans la convention de l'unité). Ce courrier demande une réponse sous quinze jours. Le directeur du département scientifique notifie son accord au département SDV.
La cellule éthique du département SDV adresse au service financier du siège une demande de paiement du « droit fixe ».
Le service financier de la délégation du siège liquide et ordonnance le droit fixe (900 F au 1er janvier 1998 pour les organismes de formation et de recherche sans but lucratif). Le paiement à la DRASS est effectué par l'ACP et le reçu attestant le paiement pour le CCPPRB est retourné au département SDV.
La cellule éthique complète l'imprimé d'attestation d'assurance. À réception du reçu et de l'accord scientifique et juridique, le directeur du département SDV signe la première page de la demande d'avis au CCPPRB, par laquelle le CNRS s'engage en tant que promoteur de la recherche. L'ensemble du dossier est retourné à l'investigateur.
et 7 bis . L' investigateur envoie le dossier complet au CCPPRB qui rend son avis (réunion mensuelle du comité). Au cas où des données nominatives devraient être informatisées, l'investigateur demande l'autorisation de la CNIL par l'intermédiaire de la DCAJ.
À réception de l'avis du CCPPRB, l' investigateur remplit la déclaration d'intention (cerfa no 65-0042 ou 65-0038, modèles disponibles sur le WEB) et les pièces annexes requises. L'ensemble des pièces est transmis au département SDV.
Le directeur du département SDV signe en tant que promoteur la déclaration d'intention qui est adressée à la direction générale de la santé (DGS) (ou à l'agence du médicament - ADM) en recommandé avec accusé de réception. L'investigateur est avisé par courrier électronique de l'accusé de réception postal. Un exemplaire de la déclaration d'intention est retourné par la DGS avec un numéro d'enregistrement. Une copie est alors adressée à l'investigateur accompagnée d'un courrier précisant les éventuelles conditions d'indemnisation des sujets.
et 10 bis . Dès transmission de l'accusé de réception postal de la déclaration d'intention, l' investigateur peut démarrer l'expérimentation telle que décrite dans la déclaration d'intention et dans la mesure où l'avis du CCPPRB était favorable. Dans le cas contraire il doit attendre deux mois à défaut d'une autorisation/suspension/interdiction du protocole par la DGS/ADM. Il complète et transmet sans tarder la déclaration de mise en oeuvre (DMO) visée par le directeur d'unité et destinée à la compagnie d'assurance, faute de quoi la recherche ne serait pas couverte.
La cellule éthique du département SDV actualise le fichier de suivi des protocoles et « récupère » éventuellement auprès des investigateurs les attestations d'assurance et accords du CNRS non utilisés. Elle informe la délégation de la mise en oeuvre du protocole par copies de la DMO et du courrier précisant les conditions d'indemnisation. Dans le cas de locaux non CNRS, elle informe également le responsable de l'établissement qui a visé l'autorisation des lieux, par copie de la DMO. Elle établit un relevé trimestriel des DMO et le transmet à la délégation du siège.
Le service financier de la délégation du siège adresse le relevé à la compagnie d'assurance.
2.3. Procédure : Mise en oeuvre du protocole de recherche biomédicale
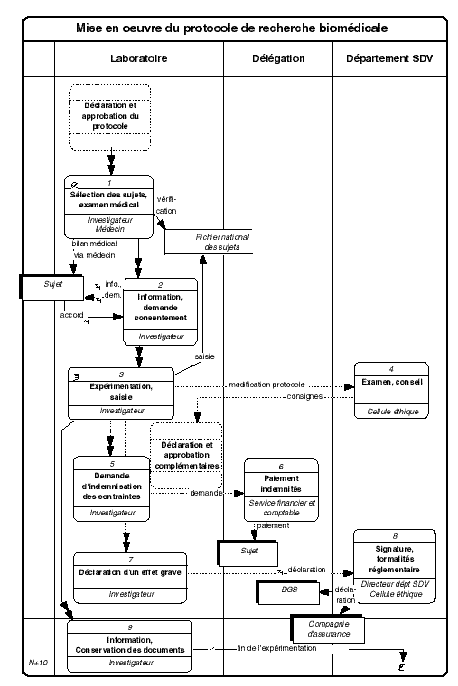
Lorsqu'il a reçu la déclaration d'intention enregistrée par la DGS, et qu'il a envoyé la déclaration de mise en oeuvre, l' investigateur peut procéder à la sélection des sujets. Dans le cas d'une recherche sans bénéfice individuel direct, il s'assure que le sujet est affilié ou qu'il bénéficie d'un régime de sécurité sociale ; qu'il n'est pas dans une période d'exclusion suite à un protocole précédent ou en cours, en consultant le fichier national des personnes qui se prêtent à des expériences biomédicales ; il fait procéder par un médecin à un examen médical de la personne, à qui les résultats sont communiqués par le médecin de son choix.
L' investigateur recueille le consentement « éclairé » du
sujet. Pour cela, il l'informe des objectifs et des conditions de la recherche, des
risques encourus et des bénéfices attendus. La personne donne son accord écrit en
cosignant avec l'investigateur, un document en deux exemplaires originaux (voir modèles
en annexes) (1). Elle reçoit l'un des exemplaires et l'investigateur conserve l'autre.
Dans certains cas, le consentement peut être recueilli auprès de proches ou de personnes
ayant la tutelle du sujet ( cf. tableau p . 45).
Lorsque dans l'intérêt d'une personne malade, le diagnostic de sa maladie n'a pu lui
être révélé, l' investigateur peut
réserver certaines informations liées à ce diagnostic. De même, l'objectif et la
méthodologie d'une recherche en psychologie peuvent n'être communiqués que de façon
succincte dans un premier temps. Ces deux dernières éventualités doivent avoir été
mentionnées dans le protocole soumis au CCPPRB.
Au fur et à mesure du déroulement du protocole, l'investigateur actualise le fichier national des sujets car nul ne peut se prêter simultanément à plus d'un protocole de recherche sans bénéfice individuel direct.
Si une modification « substantielle » du protocole (conditions du protocole, nombre de sujets, durée) doit intervenir, l'investigateur doit envoyer un projet au département des sciences de la vie. La cellule éthique du département SDV examine la modification souhaitée et le conseille sur les formalités complémentaires à accomplir : information ou demande d'avis du CCPPRB, déclaration d'intention complémentaire au ministère chargé de la santé (cerfa no 65-0043 ou 65-0039) , information de la compagnie d'assurance s'il y a augmentation du nombre de sujets et de la durée du protocole. La procédure est alors semblable à la procédure de déclaration et d'approbation initiale.
Le cas échéant, lors d'une recherche sans bénéfice individuel direct sur une personne non « protégée », l' investigateur peut proposer une indemnisation des contraintes subies par le sujet. Il adresse sa demande accompagnée des pièces justificatives à la délégation lorsque l'indemnisation s'effectue sur des crédits gérés par le CNRS ( cf. chapitre 4 et formulaire en annexe) (1).
Le service financier et comptable de la délégation verse au sujet les indemnités, au vu d'une décision signée par le délégué régional (modèle en annexe)4 .
En cas d'apparition d'un effet grave sur un sujet, l' investigateur doit prévenir rapidement le département SDV et lui adresser une déclaration complétée (cerfa no 65-0044 ou 65-0040).
Le directeur du département SDV signe la déclaration en tant que promoteur de la recherche, et l'adresse à la direction générale de la santé (ou à l'agence du médicament) ainsi qu'à la compagnie d'assurance.
Dès la fin de l'expérimentation, l' investigateur informe la cellule éthique du département SDV. Le cas échéant il complète la saisie sur le fichier national des sujets afin de mentionner les indemnités compensatrices versées et de faire courir la période d'exclusion. Tous les documents relatifs au protocole doivent être conservés pendant 10 ans. En cas de problème, fermeture de l'unité par exemple, la cellule éthique du département SDV peut être consultée.
- loi « Huriet-Sérusclat » : loi no 88-1138 du 20-12-1988 ( JO du 22-12-1988) principalement modifiée par les lois no 90-86 du 23-01-1990 ( JO du 25-01-1990), no 91-73 du 18-01-1991 ( JO du 20-01-1991) et n o 94-630 du 25-07-1994 ( JO du 26-07-1994) ;
- décret d'application du 20-09-1990.
Ces deux textes forment le livre II bis du code de la santé publique (article 209).
Recherche avec l'homme in Éthique en sciences de la vie - Guide pratique. Département des sciences de la vie du CNRS, janvier 1997 (nouvelle édition prévue en 1999).
Informatique et libertés - Guide pratique à l'usage des chercheurs. Direction des contrats et des affaires juridiques du CNRS, 1996.
Guide pratique : Loi du 20 décembre 1988 modifiée sur la protection des personnes qui se prêtent aux recherches biomédicales , Doin éditeurs/AP-HP, 1997, coll. Les guides de l'AP-HP.
4. - PRINCIPES ET RÈGLES DE GESTION APPLICABLES
Les personnes exerçant au sein des structures de recherche et de service du CNRS, quels que soient leurs statuts et leurs organismes d'appartenance, peuvent lui demander d'être promoteur de la recherche. Cet engagement n'est délivré que pour le protocole proposé.
Lorsque le protocole s'inscrit dans le cadre d'une convention de recherche, celle-ci doit mentionner quelle partie sera promoteur de la recherche. Lorsque c'est le partenaire qui est désigné, la convention doit préciser que c'est à lui de payer aux sujets les éventuelles indemnités.
La qualité de promoteur de la recherche biomédicale est distincte de celle de propriétaire des droits qui reste liée aux statuts et conventions gouvernant l'unité.
Les actes et examens médicaux spécifiquement liés au protocole de recherche, y compris les journées d'hospitalisation, sont facturés au promoteur car ils ne sont pas pris en charge par la sécurité sociale. Ils peuvent être pris en charge par le laboratoire.
Les frais supportés par le sujet (frais de transport essentiellement) peuvent être pris en charge quel que soit le type de la recherche, avec ou sans bénéfice direct.
La loi prévoit la possibilité pour le promoteur d'indemniser les sujets (sauf populations protégées cf. supra ) pour les contraintes subies dans le cadre des recherches sans bénéfice individuel direct.
- Le CNRS ne paye ces indemnités que lorsqu'il est promoteur de la recherche.
- Le CNRS promoteur de la recherche doit être informé si l'indemnisation est versée par un tiers.
- Le CNRS n'indemnise qu'au vu de l'accusé de réception de la déclaration d'intention et d'un document annonçant le nombre total de sujets et l'indemnisation prévue (la demande d'avis au CCPPRB mentionne ces informations). La délégation vérifie le non-dépassement de ce contingent.
- Un sujet ne peut recevoir au total plus de 25 000 F (au 1 er juin 1998) sur 12 mois, d'indemnités pour contraintes subies quel que soit le nombre de protocoles. L'investigateur doit s'en assurer en consultant le fichier national des sujets, et doit compléter ce dernier.
- Les indemnités perçues par les sujets sont non imposables.
- Les indemnités sont à prendre sur les crédits du laboratoire en soutien de base - code (0690), éventuellement en actions incitatives - code (0693), le compte d'exécution étant le 628.1 « concours divers ».
Possibilités de participation de ces populations aux recherches avec ou sans bénéfice individuel direct et modalités de recueil du consentement.
5. - MODÈLES DE DOCUMENTS 5
- Demande d'autorisation de lieu de recherches biomédicales
- Lettre de demande au CNRS d'être promoteur de la recherche
- Modèle de consentement pour une recherche avec bénéfice individuel direct
- Modèle de consentement pour une recherche sans bénéfice individuel direct
- Déclaration d'intention [ cerfa no 65-0042 ]
- Déclaration d'intention pour l'essai d'un médicament [ cerfa no 65-0038 ]
- Déclaration de mise en oeuvre pour la compagnie d 'assurances
- Demande d'indemnisation pour une recherche sans bénéfice individuel direct
- Décision d'indemnisation pour une recherche sans bénéfice individuel direct
- Déclaration d'intention complémentaire [ cerfa no 65-0043 ]
- Déclaration d'intention complémentaire (médicament) [ cerfa no 65-0039 ]
- Déclaration d'un effet grave survenu au cours d'une recherche [ cerfa no 65-0044 ]
- Déclaration d'un effet grave (médicament) [ cerfa no 65-0040 ]
La présente instruction sera publiée au Bulletin officiel du Centre national de la recherche scientifique.
Fait à Paris, le 30 octobre 1998.
Le secrétaire général,
Jean-Pierre SOUZY
1. Annexes non reproduites
2. Annexes non reproduites
3. Annexes non reproduites
4. Annexes non reproduites
5. Les modèles de documents sont disponibles dans la rubrique Procédures sur le site Web du secrétariat général à l'adresse suivante : http://www.sg.cnrs.fr/bpc/procedures/procedfiju/huriet/huriet.htm